Product Description
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disease of the motor system, characterized by selective and progressive loss of motor neurons, eventually leading to paralysis and death within 2–5 years [1]. iPSC-derived motor neurons are valuable tools for biochemical analysis, disease modelling and clinical application of this disease. Cytoplasmic accumulation and nuclear loss of the RNA binding protein transactive response DNA-binding protein 43 (TDP-43) from affected neurons in most instances of ALS [2-3]. Over 40 dominantly inherited mutations in the gene encoding TDP-43 have subsequently been identified in familial ALS patients [4], implicating TDP-43 dysfunction in the vast majority of ALS cases.
Human Motor Neurons (iPSC-derived, TDP-43 mutation, N352S) is derived from a genetically modified normal iPSC line carrying the heterozygous or homozygous N352S mutation in the TDP-43 gene (Figure 1). iXCells™ hiPSC-derived motor neurons express typical markers of motor neurons, e.g. HB9 (MNX1), ISL1, CHAT, (Figure 2), with the purity higher than 85%. iXCells™ motor neurons are available in both cryopreserved vials and fresh plate formats (12, 24, 48, and 96-well plate). Most of the cells will express high level of HB9 and ISL-1 after thawing in the Motor Neuron Culture Medium Kit (Cat# MD-0022-100ML). And after cultured in the medium for 5-7 days, these cells will express high levels of CHAT and MAP2.

Figure 1. Heterozygous and homozygous N352S mutation (highlighted) has been introduced to TDP-43 gene using CRISPR/Cas9 based genome editing technology. The targeted site is verified by genomic PCR/Sanger sequencing.
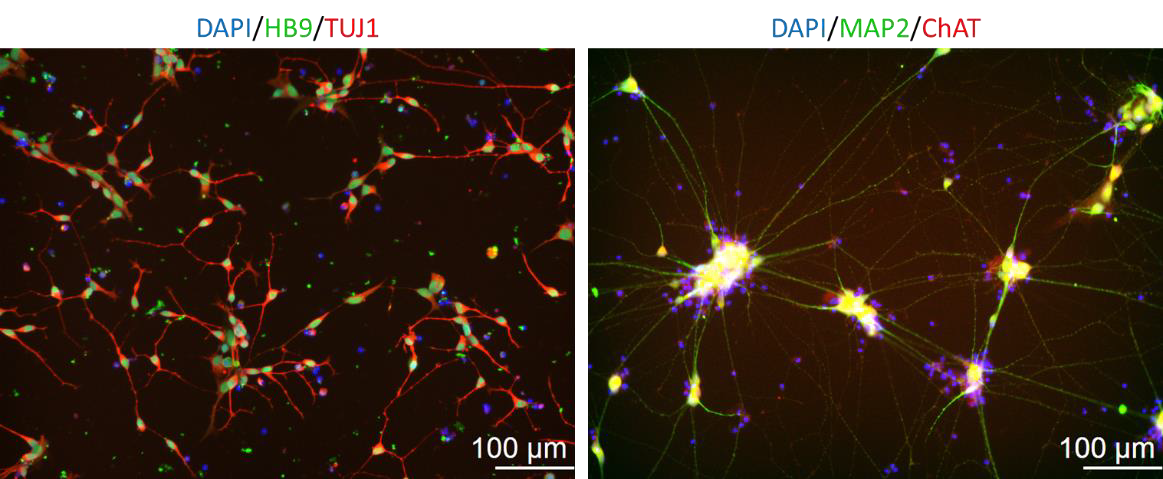
Figure 2. Immunofluorescence staining showing HB9 and ChAT positive cells on day 2 and 7 in culture respectively.
Product Details
| Organism | Homo Sapiens, Human |
| Cell Type | Brain Cell |
| Disease | ALS |
| Package Size | 1 x 106 cells/vial and 2 x 106 cells/vial |
| Passage Number | P0 |
| Growth Properties | Adherent |
| Shipped | Cryopreserved |
| Storage | Liquid Nitrogen |
| Associated Media | iXCells™ Motor Neuron Culture Medium Kit (Cat# MD-0022-100ML) |
References
[1] Taylor, J. P., Brown, R. H. Jr & Cleveland, D. W. Decoding ALS: from genes to mechanism. Nature 539, 197–206 (2016).
[2] Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
[3] Ling, S. C., Polymenidou, M. & Cleveland, D. W. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 (2013).
[4] Lagier-Tourenne, C., Polymenidou, M. & Cleveland, D. W. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64 (2010).