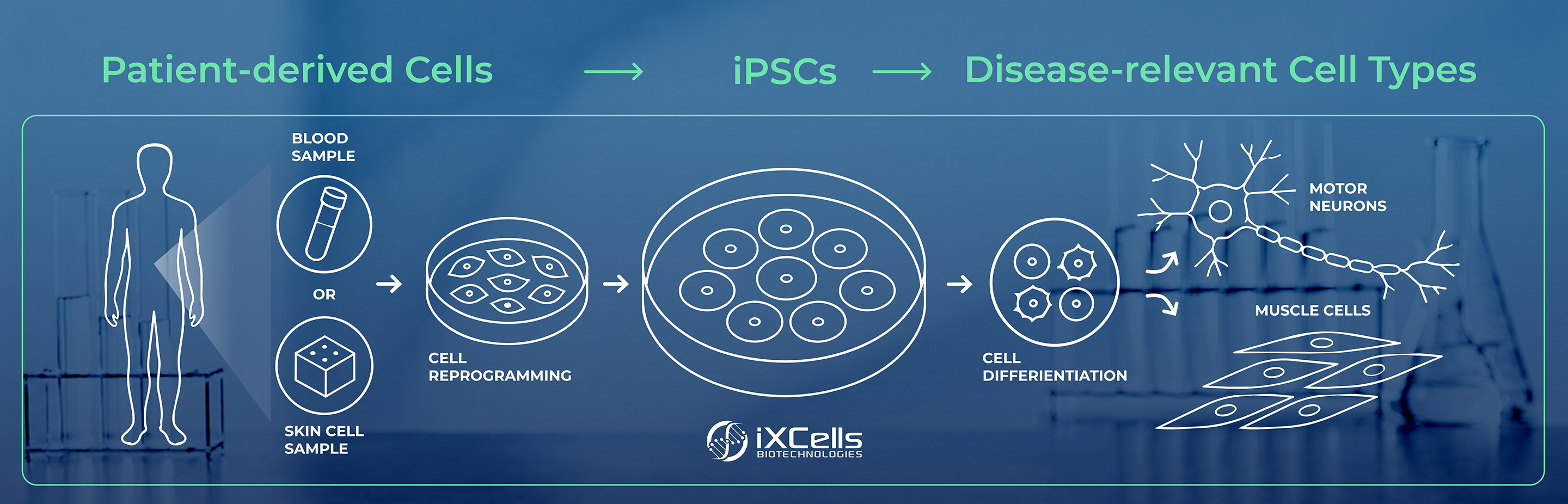
Induced pluripotent stem cell-derived models are helping researchers study rare diseases in more human-relevant, patient-specific ways. In this article, we look at how iPSC-based disease models have been used in ALS and Duchenne muscular dystrophy research to investigate disease mechanisms, study mutation-specific effects, and support early-stage research in vitro.
iPSC models are used to create patient-specific cellular systems that help researchers study disease mechanisms, mutation-specific effects, and early research questions in vitro.
They allow scientists to investigate rare diseases in human cells that more closely reflect the genetic background of disease than many traditional model systems.
This article highlights case studies in ALS and Duchenne muscular dystrophy to show how iPSC-derived models support rare disease research, mechanistic studies, and early preclinical investigation.
Rare diseases present some of the most difficult challenges in biomedical research. Many are driven by specific genetic abnormalities, involve complex cellular pathology, and affect tissues that are not easily accessible for direct study. In many cases, researchers also face a lack of representative model systems that fully capture the biology of disease in a human context.
Induced pluripotent stem cells, or iPSCs, have become an important tool in this area of research because they can be used to generate patient-specific disease models in the lab. In this context, the focus is not on using iPSCs as a replacement therapy, but on using iPSC-derived cells as research models that help scientists study rare disease biology in vitro.¹˒²
For researchers working in rare disease biology, this matters for a simple reason: the closer a model comes to reflecting the disease as it appears in human cells, the more useful it can be for studying mechanisms, evaluating hypotheses, and informing early preclinical research.
In this article, we highlight several targeted applications of iPSC-derived models in rare disease research. Through case studies in amyotrophic lateral sclerosis (ALS) and Duchenne muscular dystrophy (DMD), we explore how iPSC-based models have helped researchers investigate disease-associated phenotypes, better understand mechanisms of pathology, and support the evaluation of early research strategies in vitro.¹˒²
Rare diseases often involve highly specific genetic drivers, which makes patient-derived models especially valuable. iPSC technology allows researchers to start with adult cells collected from a patient, reprogram them into a pluripotent state, and then differentiate them into disease-relevant cell types for in vitro study.¹˒²
This approach offers several important advantages for rare disease research. First, iPSC-derived models provide a more human-relevant system than many traditional animal models, allowing researchers to study disease biology in human cells that retain the patient’s genetic background. This makes it possible to examine how specific mutations or variants may influence cellular behavior in a disease-relevant context.
Second, iPSC-derived models can provide access to cell types that may otherwise be difficult or impossible to obtain directly from patients, such as motor neurons, astrocytes, cardiomyocytes, or muscle cells. This is especially important in rare diseases that affect tissues that are not easily accessible for direct study.
Third, iPSC-derived models are highly scalable. As differentiation, assay design, and automation workflows continue to improve, these models can support larger-scale studies, including high-content imaging and screening approaches used in early drug discovery and development.
iPSC-derived models also create opportunities to study more complex biology through co-culture systems, organ-on-chip platforms, and other multi-cell-type models. Because different disease-relevant cell types can be generated from the same iPSC disease background, researchers can investigate interactions between cells that may contribute to disease progression, including non-cell-autonomous events where one cell type influences the function or survival of another.
Over the past decade, iPSC-derived models have become increasingly important in rare disease research because they support a more direct investigation of pathology in human cells. As with any model system, they have limitations and must be interpreted within the broader context of preclinical research. But for many rare diseases, they offer an important platform for studying biology in a way that is human-relevant, patient-specific, scalable, and experimentally tractable.¹
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease marked by the loss of motor neurons. In many forms of ALS, one of the most important pathological features is abnormal behavior of the RNA-binding protein TDP-43 in affected cells.³
To better understand how this pathology develops, researchers have generated iPSCs from ALS patients and differentiated them into motor neurons. In one notable study, these patient-derived motor neurons reproduced aspects of TDP-43-associated pathology observed in disease, helping researchers examine cellular vulnerability in a more disease-relevant setting.³
This type of model is significant because it allows investigators to study pathogenic features directly in human motor neurons carrying disease-associated genetic backgrounds. Instead of inferring disease mechanisms solely from nonhuman systems or non-neuronal cell types, researchers can observe how pathology emerges in cells that are more directly connected to the affected tissue.³
The broader value of this work lies in what the model makes possible. iPSC-derived motor neurons can help researchers investigate how TDP-43 dysfunction contributes to neurodegeneration, identify cellular phenotypes associated with disease progression, and establish a platform for evaluating early research hypotheses in vitro.³
For rare neurodegenerative diseases, that kind of human-relevant model can provide a more informative starting point for mechanistic research and early-stage preclinical exploration.
A second important application of iPSC-derived models in ALS research involves mutation-specific modeling. In one study, researchers focused on mutations in the SOD1 gene, which are associated with certain familial forms of ALS. The same study also included C9orf72 mutation lines, adding another important genetic context for examining ALS biology.⁴
SOD1 mutations have been linked to motor neuron toxicity and disrupted cellular pathways, making them an important focus in efforts to better understand ALS disease mechanisms. Using iPSCs derived from patients with SOD1 mutations, researchers developed motor neuron models that captured features of disease-associated dysfunction in vitro. These models helped investigators study how SOD1-related pathology affects neuronal health and how specific molecular pathways may contribute to degeneration.⁴
One of the major strengths of this research is the ability to examine mutation-specific effects in a controlled setting. In studies using genetically corrected comparison lines, researchers have been able to distinguish disease-related phenotypes from background variation more clearly. This kind of design strengthens mechanistic interpretation by allowing scientists to connect a specific mutation more directly to an observed cellular outcome.⁴
In practical terms, SOD1 iPSC models have contributed to a better understanding of how disease-associated genetic alterations influence motor neuron biology. The inclusion of C9orf72 mutation lines also underscores the broader value of iPSC-derived models for studying ALS across different genetic backgrounds. Together, these approaches support research into genotype-to-phenotype relationships in a system that is more closely aligned with human disease than many traditional preclinical models.⁴
As iPSC workflows continue to improve, models like these are becoming increasingly useful for studying mutation-specific biology in rare neurodegenerative disorders.
Duchenne muscular dystrophy is a rare genetic disorder caused by mutations in the dystrophin gene. The disease leads to progressive muscle degeneration and has long been a focus of research aimed at understanding how loss of dystrophin affects muscle structure and function.
iPSC technology has provided researchers with a way to model DMD using patient-derived cells that can be differentiated into muscle-relevant cell types. These models make it possible to study disease-associated phenotypes in a human genetic context and to assess how cellular function changes when dystrophin is absent or disrupted.⁵
This has been particularly valuable in research exploring gene-editing strategies. In preclinical studies, investigators have used CRISPR-Cas9-based approaches in hiPSC-derived muscle cells to evaluate whether targeted correction of dystrophin-related mutations can restore key aspects of normal cellular function.⁵
iPSC-derived DMD models have also been used to evaluate other mutation-directed approaches, including exon-skipping strategies and base-editing methods designed to restore or improve dystrophin expression in patient-derived cells.⁶˒⁷
The importance of this work is not only in demonstrating proof of concept, but in showing how iPSC-derived disease models can support therapeutic testing earlier in development. By giving researchers access to patient-specific cellular systems, these models can help refine experimental questions, evaluate editing strategies, and generate data that may inform the next stages of preclinical research.
Taken together, these examples illustrate why iPSC-derived models have become such an important tool in rare disease research. Their value is not simply that they can generate disease-relevant cell types in the lab. It is that they give researchers a way to study pathology in models that retain meaningful aspects of the patient’s genetic context.¹˒²
In ALS, iPSC-derived motor neurons have helped researchers study disease-associated protein pathology and mutation-linked dysfunction in a more targeted way.³˒⁴ In DMD, iPSC-derived muscle models have supported the study of dystrophin-related defects and the evaluation of gene-editing approaches in human cells.⁵
These kinds of applications are especially important in rare disease research, where limited patient access and limited model availability have historically slowed progress. iPSC-derived models do not eliminate those challenges entirely, but they do provide a more flexible and biologically relevant platform for addressing them.¹
Just as importantly, they fit well within a broader translational research workflow. iPSC models can contribute to early mechanistic studies, support target validation, and help researchers evaluate experimental concepts before moving into more advanced stages of development.¹˒²
The role of iPSC-derived models in rare disease research will likely continue to grow as reprogramming, differentiation, genome editing, and assay design become more refined. As these workflows improve, researchers will have even greater ability to generate disease-relevant models that support more precise and informative studies.¹˒²
For rare diseases in particular, that progress matters. These conditions often require highly tailored approaches to understanding pathology and evaluating potential interventions. iPSC-derived disease models are well suited to that challenge because they enable the development of patient-specific cellular systems that can illuminate disease biology in a more human-relevant framework.¹
While no single model system can answer every question, iPSC-derived models have clearly expanded what is possible in rare disease research. They are helping researchers move closer to models that are not only more personalized, but also more useful for connecting mechanism, phenotype, and research strategy.¹˒²
iPSC-derived models have become a valuable part of rare disease research by enabling patient-specific systems that can capture important aspects of disease biology in vitro. Through applications in ALS, DMD, and other genetically defined disorders, they have helped researchers study pathology more directly, investigate mutation-specific effects, and support early preclinical research.¹˒³˒⁴˒⁵
Their growing importance lies in their ability to bring greater biological relevance to rare disease research. By making it possible to study disease-associated genetic backgrounds in human cells, iPSC models help researchers ask better questions and generate more informative data.¹˒²
As the field continues to evolve, iPSC-derived disease models will remain an important tool for advancing the study of rare diseases and strengthening the connection between early discovery and translational research.¹˒²
Author:
Dr. Xinyu Kong, PhD
Director of iPSC Generation and Gene Editing
iXCells Biotechnologies
With over a decade of experience in cellular biology and preclinical research, iXCells delivers human-relevant models that help researchers improve predictability and move new therapies forward.